Literature Review: Molecular Property Prediction
Molecular Property Prediction is a fundamental cheminformatic task in the field of drug discovery. The accurate prediction of molecular properties can significantly accelerate the overall process of finding drug candidates in a faster and cheaper way. - [1]
Deep Learning for Molecular Property Prediction has been actively researched with focus on 2 molecular representations: Simplified Molecular Input Line Entry System (SMILES) and Graph Representation
Simplified Molecular Input Line Entry System (SMILES)
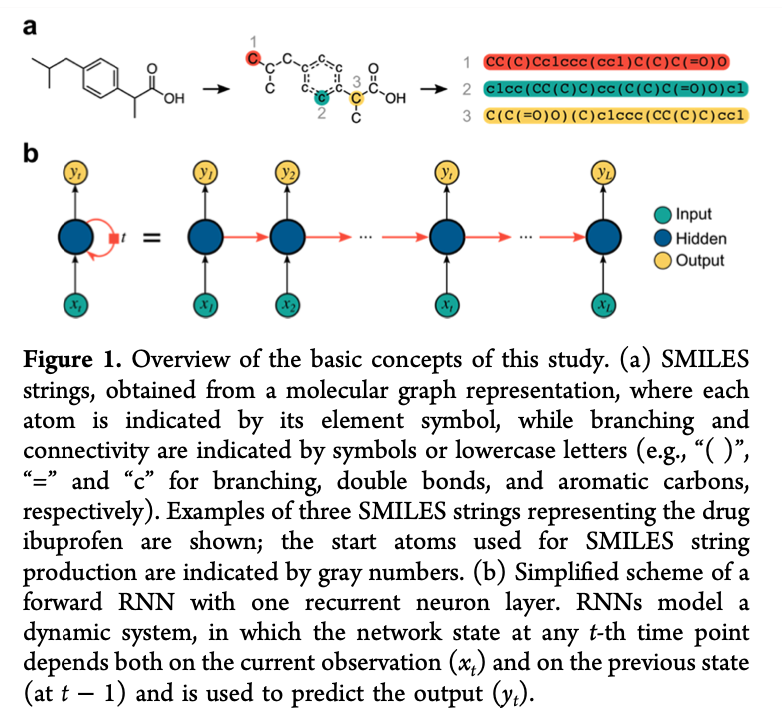
SMILES is a chemical notation that can fully convert molecular structures into a linear format that can be used by the computer [2, 66].
Research work in [3, 4, 5, 7, 8, 9] has utilized Recurrent Neural Networks (RNNs) to extract features from the SMILES strings of molecules to predict molecular properties. Instead of formulating this problem as a simple regression task, they developed molecular generative models which performed two tasks: reconstructing the SMILES strings and predicting molecular properties. The reconstruction objective was proven to improve the property prediction accuracy. A potential drawback of the character-by-character SMILES and other linear formats is the reconstruction of invalid molecules which could lead to the property prediction error [10].
Look at the figure below, (a) shows the processing of decoding molecular graphs into SMILES strings and (b) shows the RNNs applied on SMILES strings. Vanishing gradient is the major limit of RNN variants that little or no gradients will be updated for last characters of long SMILES strings. As a result, RNNs may not handle huge molecule graphs.
Graph Representation
Variants of Graph Neural Networks have been introduced to take input different structure levels structure levels (i.e. atoms, motifs) [1, 11, 12, 14] and to predict molecular properties while reconstructing original molecule graphs. These two tasks are discussed in details in the next section.
Similar to SMILES, each node (aka atom) or edge (aka bond) has its distinct embedding. In the linearly-ordered SMILES, RNNs update hidden states of the next atom from the previous atom. However, in graph, each atom has no information of its neighbor atoms that requires Message Passing Neural Networks (MPNN) to update hidden states of atoms and edges by collecting information of neighbor atoms and edges in T iterations [1, 11, 12, 14].
In the figure below, we assume that MPNN propagates in 2 iterations. In the 1st iteration, atom 2 is updated by three direct neighbor atoms 1, 3, and 4; and atom 1 is directly updated from atoms 2 and 5. In the 2nd iteration, atom 2 which carries information of atom 5 is used to update atom 2. Similarly, when increasing T iterations, more indirect atoms are utilized to update the current atom.
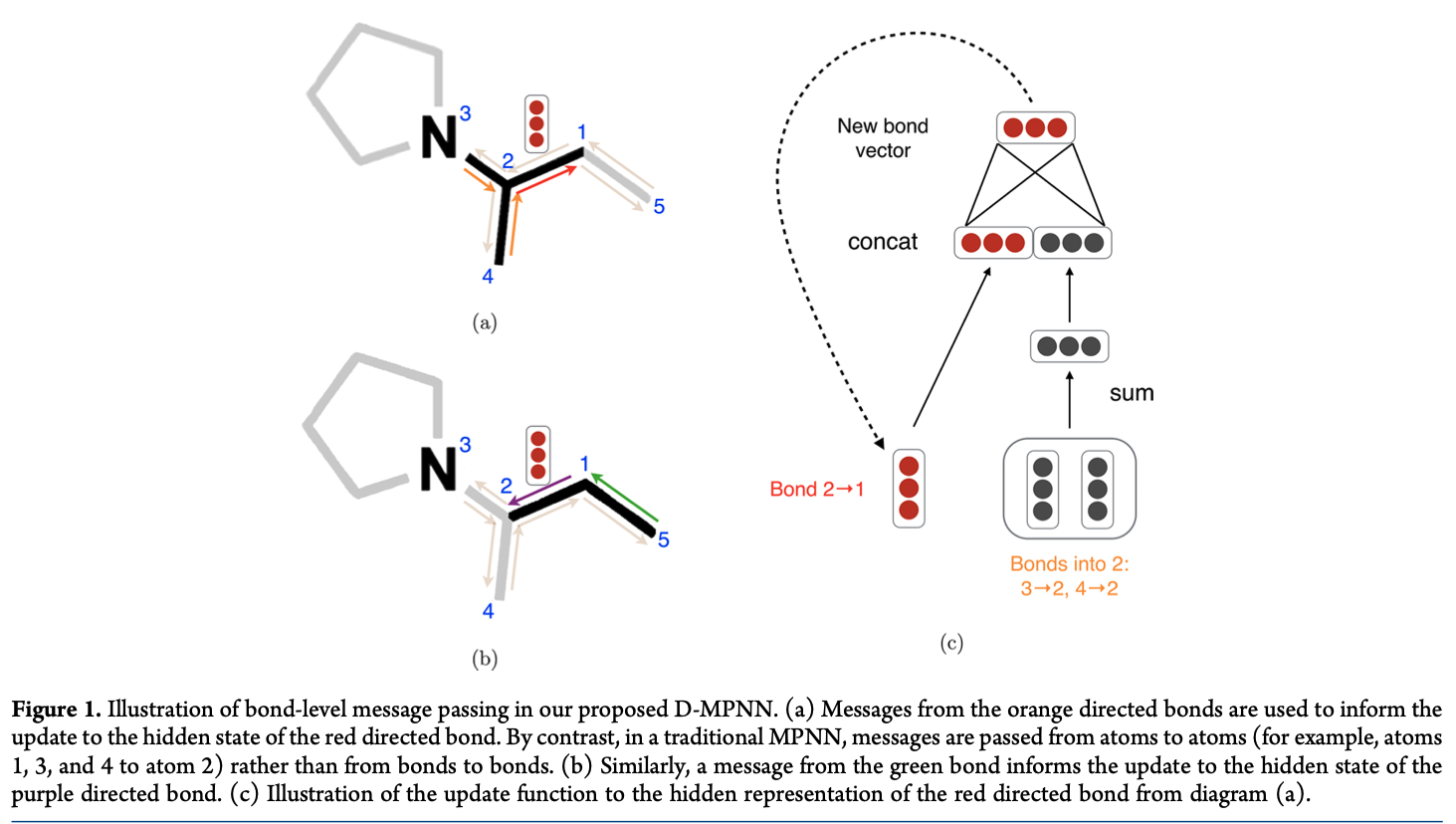
After T iterations, the hidden states representing the graph is computed from hidden states of all atoms and edges (usually summation) and is fed to the output model for property prediction
Molecular Graph-based Property Prediction
Graph-based property prediction is actively researched with focus on two settings: with and without the graph reconstruction task [11].
Without Graph Generation
In this setting, the encoder architecture including MPNNs and the output model performs the property prediction task only. In the figure below, without the graph reconstruction task, the output from the encoder could be directly used to predict molecular properties.
With Graph Generation
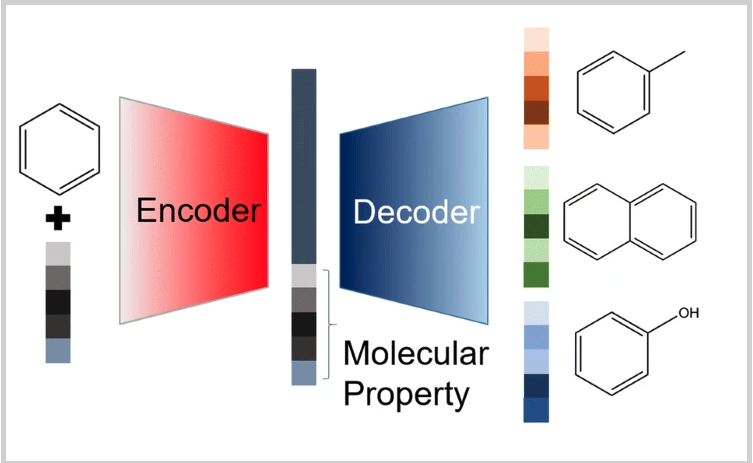
In this setting, the Encoder-Decoder (aka AutoEncoder) architecture including MPNNs and a graph generator performs the graph reconstruction task [11, 12, 14]. Some models are trained and predicted in the autoregressive mode which inherits from the seq2seq modeling in Natural Language Processing [12, 14]. Besides, an additional model predicts properties given input from latent representations generated by the encoder [10, 14]. The multi-task setting has been actively experimented and reported to improve the molecular property prediction.
The figure below shows the AutoEncoder architecture performing the graph reconstruction and property prediction tasks.
References
[1] - A compact review of molecularproperty prediction with graph neural networks
[2] - SMILES Tutorial
[3] - Randomized SMILES strings improve the quality of molecular generative models
[4] - Bidirectional Molecular Generation with Recurrent Neural Networks
[5] - Generating Focused Molecule Libraries for Drug Discovery with Recurrent Neural Networks
[6] - SMILES, a Chemical Language and Information System
[7] - In Silico Generation of Novel, Drug-like Chemical Matter Using the LSTM Neural Network
[8] - Generative Recurrent Networks for De Novo Drug Design
[9] - Molecular Generation with Recurrent Neural Networks (RNNs)
[10] - Automatic Chemical Design Using a Data-Driven Continuous Representation of Molecules
[11] - Analyzing Learned Molecular Representations for Property Prediction
[12] - Molecular generative model based on conditional variational autoencoder for de novo molecular design
[14] - Hierarchical Generation of Molecular Graphs using Structural Motifs